سینتیک شاخه ای از شیمی فیزیک است که به دو دسته ماکروسینتیک و سینتیک مولکولی تقسیم می شود . سینتیک مولکولی به زمانبندی واکنشهای شیمیایی در سطح مولکولی میپردازد و شامل میکروسینتیک است که به سینتیک واکنشهای ابتدایی میپردازد. ماکروسینتیک تأثیر فرآیندهای انتقال حرارت و جرم ماکروسکوپی را بر سینتیک واکنشهای شیمیایی در نظر میگیرد و بنابراین نشاندهنده ارتباط بین سینتیک واکنش و فناوری واکنش شیمیایی است. این مقاله به سینتیک مولکولی واکنشهای شیمیایی میپردازد. سینتیک فرآیندهای الکتروشیمیایی در مقاله سینتیک الکتروشیمیایی مورد بحث قرار گرفته است .
سرعت واکنش
تعریف

 ، اشتقاق آند([آ]/[آ]0)/دتی
، اشتقاق آند([آ]/[آ]0)/دتی و سرعت واکنش1/�آ⋅د([آ]/[آ]0)/دتی
و سرعت واکنش1/�آ⋅د([آ]/[آ]0)/دتی به عنوان تابعی از زمان واکنشتی
به عنوان تابعی از زمان واکنشتی .[آ]0
.[آ]0 غلظت اولیه است،[آ](تی)
غلظت اولیه است،[آ](تی) غلظت با توجه به زمان واکنشتی
غلظت با توجه به زمان واکنشتی عدد استوکیومتری مواد اولیهآ
عدد استوکیومتری مواد اولیهآ .[آ](تی)/[آ]0
.[آ](تی)/[آ]0 غلظت نسبی بدون بعد استآ
غلظت نسبی بدون بعد استآکمیت اصلی سینتیک سرعت واکنش است .�با ابعاد کمیت ماده در هر زمان و حجم . [2] [3] واحد سرعت واکنش به دست آمده از کمیت های اساسی سیستم بین المللی واحدها مول بر متر مکعب و ثانیه است . نرخ واکنش نشان دهنده تغییر متغیر فروش است �(میزان رویدادهای واکنش، که با معادله واکنش واکنش مورد بررسی، بر حسب مول تعریف می شود) در واحد زمان و حجم در شرایط ایزوکوریک . هستند�منعدد استوکیومتری و|�من|مقدار عدد استوکیومتری یک ماده درگیر در واکنش مورد بررسیمن، واکنش نشان خواهد داد
 با ابعاد کمیت ماده در هر زمان و حجم . [2] [3] واحد سرعت واکنش به دست آمده از کمیت های اساسی سیستم بین المللی واحدها مول بر متر مکعب و ثانیه است . نرخ واکنش نشان دهنده تغییر متغیر فروش است �
با ابعاد کمیت ماده در هر زمان و حجم . [2] [3] واحد سرعت واکنش به دست آمده از کمیت های اساسی سیستم بین المللی واحدها مول بر متر مکعب و ثانیه است . نرخ واکنش نشان دهنده تغییر متغیر فروش است � (میزان رویدادهای واکنش، که با معادله واکنش واکنش مورد بررسی، بر حسب مول تعریف می شود) در واحد زمان و حجم در شرایط ایزوکوریک . هستند�من
(میزان رویدادهای واکنش، که با معادله واکنش واکنش مورد بررسی، بر حسب مول تعریف می شود) در واحد زمان و حجم در شرایط ایزوکوریک . هستند�من عدد استوکیومتری و|�من|
عدد استوکیومتری و|�من| مقدار عدد استوکیومتری یک ماده درگیر در واکنش مورد بررسیمن
مقدار عدد استوکیومتری یک ماده درگیر در واکنش مورد بررسیمن ، واکنش نشان خواهد داد
، واکنش نشان خواهد داد- |�آ|آ+|�ب|ب+… ⟶ |�ک|ک+|�L|L+…

سرعت واکنش�زوج:
- �=د��دتی=1�آد[آ]دتی=1�بد[ب]دتی=1�کد[ک]دتی=1�Lد[L]دتی

اینجا هستندتیزمان واکنش،�حجم واکنش و[آ]،[ب]،[ک]همچنین [L]غلظت مواد مرتبط با حجم مواد درگیر در واکنش مورد بررسیآ،ب،کوL. ضریب دیفرانسیلد[آ]/دتیبرابر با شیب نمایه غلظت-زمان زیرین است[آ](تی)، که[آ]به عنوان تابعی ازتینشان می دهد. آنجاآبه عنوان ماده اولیه مصرف می شود تغییر تفاضلی در غلظت استد[آ]و در نتیجه ضریب دیفرانسیلد[آ]/دتیمنفی. از آنجایی که عدد استوکیومتری یک ماده اولیه نیز طبق قرارداد دارای علامت منفی است، عبارت1/�آ⋅د[آ]/دتیو در نتیجه سرعت واکنش�مثبت
 حجم واکنش و[آ]
حجم واکنش و[آ] ،[ب]
،[ب] ،[ک]
،[ک] همچنین [L]
همچنین [L] غلظت مواد مرتبط با حجم مواد درگیر در واکنش مورد بررسیآ
غلظت مواد مرتبط با حجم مواد درگیر در واکنش مورد بررسیآ ،ب
،ب ،ک
،ک وL
وL . ضریب دیفرانسیلد[آ]/دتی
. ضریب دیفرانسیلد[آ]/دتی برابر با شیب نمایه غلظت-زمان زیرین است[آ](تی)
برابر با شیب نمایه غلظت-زمان زیرین است[آ](تی) و در نتیجه ضریب دیفرانسیلد[آ]/دتی
و در نتیجه ضریب دیفرانسیلد[آ]/دتی و در نتیجه سرعت واکنش�
و در نتیجه سرعت واکنش�قوانین سرعت و قوانین زمان
قوانین سرعت
وابستگی سرعت واکنش به غلظتهای فعلی واکنشدهندهها در یک واکنش بهطور تجربی با قوانین سرعت توصیف میشود. قوانین سرعت معمولاً دارای یک ثابت سرعت هستند کیا نیمه عمر تی1/2، که سینتیک فرآیند شیمیایی مورد بررسی را به روشی مشخص نشان می دهند. نیمه عمر دوره ای را نشان می دهد که در آن غلظت اولیه[آ]0یک واکنش دهندهآبه نصف مقدار کاهش می یابد.
 یا نیمه عمر تی1/2
یا نیمه عمر تی1/2 ، که سینتیک فرآیند شیمیایی مورد بررسی را به روشی مشخص نشان می دهند. نیمه عمر دوره ای را نشان می دهد که در آن غلظت اولیه[آ]0
، که سینتیک فرآیند شیمیایی مورد بررسی را به روشی مشخص نشان می دهند. نیمه عمر دوره ای را نشان می دهد که در آن غلظت اولیه[آ]0 یک واکنش دهندهآ
یک واکنش دهندهآواکنشهای ناخالص قابل مشاهده پدیدارشناسی میتوانند مکانیسمهای واکنش پیچیدهای داشته باشند که شامل توالیهایی از چندین واکنش اولیه برگشتپذیر است. نمونههایی از این واکنشهایی هستند که از مکانیسم لیندمان پیروی میکنند ، واکنشهای زنجیرهای یا واکنشهای کاتالیزشده با آنزیم که میتوانند توسط نظریه Michaelis-Menten توصیف شوند . علاوه بر این، سرعت واکنش قابل مشاهده را می توان تحت تأثیر واکنش های رقابتی قرار داد. در مورد واکنش های پلیمریزاسیون رادیکال، یک خود شتابی به نام اثر ترومسدورف می تواند در تبدیل های بالا رخ دهد . افزایش ویسکوزیته مخلوط واکنش در تبدیل های بالا و در نتیجه درجات پلیمریزاسیون بالا منجر به کاهش قابل توجهی در احتمال واکنش های خاتمه زنجیره به دلیل ترکیب مجدد انتهای زنجیره می شود، در حالی که واکنش رشد زنجیر گرمازا، که شامل افزودن مونومر به انتهای زنجیره، به دلیل افزایش تعداد انتهای زنجیره فعال، شتاب می گیرد. افزایش دمای حاصل در مخلوط واکنش به نوبه خود باعث افزایش سرعت واکنش رشد زنجیره ای می شود. از آنجایی که قوانین سرعت معمولاً رویدادهای واکنش پیچیده را نشان می دهند، مکانیسم واکنش را نمی توان مستقیماً از آنها استنباط کرد. قوانین نرخ اغلب بر اساس مدل های واکنش ساده شده فرموله می شوند. بنابراین، سینتیک واکنشهای ترکیبی که شامل چندین واکنش ابتدایی متوالی است، میتواند تحت تأثیر یک واکنش ابتدایی بهویژه آهسته به عنوان مرحله تعیینکننده سرعت باشد. در این مورد، سینتیک واکنش ترکیبی اغلب به طور رضایت بخشی توسط سینتیک ساده تر واکنش ابتدایی به خصوص کند نشان داده می شود. اگر واسطههای واکنشی در جریان واکنشها رخ دهند، میتوان از اصل شبه ایستایی بودنشتاین استفاده کرد.

قوانین زمان
قوانین زمان یا معادلات سرعت متغیر تبدیل یک واکنش یا غلظت یک ماده درگیر در یک واکنش را به عنوان تابعی از زمان واکنش مشخص می کنند. در برخی موارد، قوانین زمان واکنش های شیمیایی را می توان از قوانین سرعت مربوطه با جداسازی و ادغام متغیر تعیین کرد. برعکس، قوانین سرعت از اولین مشتقات قوانین زمان مربوطه به دست می آیند. نیمه عمر را می توان مستقیماً از قوانین زمان تعیین کرد.
وابستگی سرعت واکنش به دما
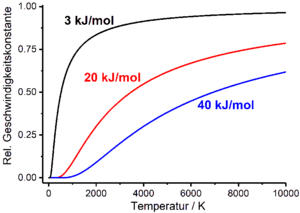
 به اشتراک گذاشته شود. دومی و انرژی فعال سازی مستقل از دما فرض می شوند.
به اشتراک گذاشته شود. دومی و انرژی فعال سازی مستقل از دما فرض می شوند.نرخ واکنش اساساً به متغیرهای حالتی که سیستم واکنش دهنده را مشخص می کند، مانند دما بستگی دارد تی، فشار و حجم. این وابستگی از طریق ثابت سرعت یا نیمه عمر، که به نوبه خود تابع دما، فشار و حجم هستند، در قوانین سرعت فرآیندهای شیمیایی گنجانده می شود. آنچه در عمل به ویژه مرتبط است، وابستگی دمایی سرعت واکنش است. این به طور تجربی توسط معادله آرنیوس توصیف میشود که ثابت سرعت را به دما مرتبط میکند. معادله آرنیوس یک تابع نمایی است که دارای یک عامل پیش نمایی استآبا بعد ثابت نرخکو انرژی فعال شدن مولی �آدر توان به عنوان پارامترهای تجربی (آرثابت عمومی گاز است ):
 ، فشار و حجم. این وابستگی از طریق ثابت سرعت یا نیمه عمر، که به نوبه خود تابع دما، فشار و حجم هستند، در قوانین سرعت فرآیندهای شیمیایی گنجانده می شود. آنچه در عمل به ویژه مرتبط است، وابستگی دمایی سرعت واکنش است. این به طور تجربی توسط معادله آرنیوس توصیف میشود که ثابت سرعت را به دما مرتبط میکند. معادله آرنیوس یک تابع نمایی است که دارای یک عامل پیش نمایی استآ
، فشار و حجم. این وابستگی از طریق ثابت سرعت یا نیمه عمر، که به نوبه خود تابع دما، فشار و حجم هستند، در قوانین سرعت فرآیندهای شیمیایی گنجانده می شود. آنچه در عمل به ویژه مرتبط است، وابستگی دمایی سرعت واکنش است. این به طور تجربی توسط معادله آرنیوس توصیف میشود که ثابت سرعت را به دما مرتبط میکند. معادله آرنیوس یک تابع نمایی است که دارای یک عامل پیش نمایی استآ و انرژی فعال شدن مولی �آ
و انرژی فعال شدن مولی �آ در توان به عنوان پارامترهای تجربی (آر
در توان به عنوان پارامترهای تجربی (آر ثابت عمومی گاز است ):
ثابت عمومی گاز است ):- ک=آ⋅ه-�آآر⋅تی

اگر به طور تقریبی فرض کنیم که ضریب پیش نمایی معادله آرنیوس و انرژی فعال سازی مستقل از دما هستند، ثابت سرعت زمانی که دما به صفر نزدیک می شود به صفر نزدیک می شود و زمانی که دما به بی نهایت نزدیک می شود در مقابل عامل پیش نمایی می شود. بنابراین ضریب پیش نمایی حداکثر مقداری را نشان می دهد که ثابت سرعت می تواند فرض کند.
به طور مشابه، از معادله آرنیوس نیز می توان برای نشان دادن اینکه نیمه عمر بستگی به دما دارد استفاده کرد. معادله آرنیوس دارای یک توان با علامت مثبت است:
- تی1/2=آ”⋅ه�آآر⋅تی

عامل پیش نمایی این را داردآ”مانند نیمه عمر بعد زمان. نیمه عمر زمانی که دما به صفر نزدیک می شود به بی نهایت نزدیک می شود و زمانی که دما به بی نهایت نزدیک می شود در مقابل عامل پیش نمایی. بنابراین ضریب پیش نمایی حداقل مقداری را نشان می دهد که نیمه عمر می تواند در نظر گرفته شود.
 مانند نیمه عمر بعد زمان. نیمه عمر زمانی که دما به صفر نزدیک می شود به بی نهایت نزدیک می شود و زمانی که دما به بی نهایت نزدیک می شود در مقابل عامل پیش نمایی. بنابراین ضریب پیش نمایی حداقل مقداری را نشان می دهد که نیمه عمر می تواند در نظر گرفته شود.
مانند نیمه عمر بعد زمان. نیمه عمر زمانی که دما به صفر نزدیک می شود به بی نهایت نزدیک می شود و زمانی که دما به بی نهایت نزدیک می شود در مقابل عامل پیش نمایی. بنابراین ضریب پیش نمایی حداقل مقداری را نشان می دهد که نیمه عمر می تواند در نظر گرفته شود.حالات گذار و انرژی های فعال سازی
حالات انتقالی
در جریان یک رویداد واکنش ابتدایی، سیستم واکنش دهنده مسیری را بر روی یک ابر سطح بالقوه طی می کند که با تغییرات ساختاری متوالی مانند تغییر در زوایای پیوند و طول پیوند مشخص می شود. با توجه به تئوری حالت گذار، یک دیوار بالقوه (سد فعال سازی) که مواد اولیه و محصولات را از هم جدا می کند، غلبه می کند، که نشان دهنده یک نقطه زین روی ابرسطح بالقوه است. حالتهایی که سیستم واکنشدهنده در طول واکنش اولیه مورد بررسی از آن عبور میکند، به راحتی با پتانسیل ترمودینامیکی مورد استفاده توصیف میشوند ، که منعکسکننده تغییرات آنتروپی در جهان ناشی از تغییرات در سیستم واکنشدهنده است. اگر فشار و دما ثابت نگه داشته شوند، این انرژی آزاد است . نقطه زین محل بالاترین انرژی آزاد است که سیستم واکنش دهنده در جریان یک رویداد واکنش اولیه از آن عبور می کند. حالتی که سیستم واکنش دهنده هنگام عبور از نقطه زینی به خود می گیرد، حالت گذار نامیده می شود . آنتالپی آزاد مولی فعال سازیΔجیاچمنn‡نشان دهنده ارتفاع مانع بالقوه ای است که برای تبدیل مواد اولیه به محصولات باید بر آن غلبه کرد، یعنی تفاوت در انرژی های آزاد حالت گذار و حالت اولیه قبل از شروع رویداد واکنش اولیه. اندازه ثابت نرخکاچمنn، که نشان دهنده سینتیک تبدیل مواد اولیه به محصولات است، بستگی داردΔجیاچمنn‡از (به بخش ” فرمولاسیون ترمودینامیکی ” در مقاله “نظریه حالت گذار” مراجعه کنید):
 نشان دهنده ارتفاع مانع بالقوه ای است که برای تبدیل مواد اولیه به محصولات باید بر آن غلبه کرد، یعنی تفاوت در انرژی های آزاد حالت گذار و حالت اولیه قبل از شروع رویداد واکنش اولیه. اندازه ثابت نرخکاچمنn
نشان دهنده ارتفاع مانع بالقوه ای است که برای تبدیل مواد اولیه به محصولات باید بر آن غلبه کرد، یعنی تفاوت در انرژی های آزاد حالت گذار و حالت اولیه قبل از شروع رویداد واکنش اولیه. اندازه ثابت نرخکاچمنn ، که نشان دهنده سینتیک تبدیل مواد اولیه به محصولات است، بستگی داردΔجیاچمنn‡
، که نشان دهنده سینتیک تبدیل مواد اولیه به محصولات است، بستگی داردΔجیاچمنn‡- کاچمنn=ثابت⋅انقضا[-Δجیاچمنn‡آرتی]

همین امر در مورد وابستگی ثابت سرعت نیز صدق می کندکrتوهجکواکنش معکوس به آنتالپی آزاد مولی فعال شدن آنها بستگی داردΔجیrتوهجک‡:
 واکنش معکوس به آنتالپی آزاد مولی فعال شدن آنها بستگی داردΔجیrتوهجک‡
واکنش معکوس به آنتالپی آزاد مولی فعال شدن آنها بستگی داردΔجیrتوهجک‡ :
:- کrتوهجک=ثابت⋅انقضا[-Δجیrتوهجک‡آرتی]

آنتالپی آزاد فعال سازی و تعادل ترمودینامیکی

 (آنتالپی آزاد واکنش).
(آنتالپی آزاد واکنش).بسیاری از واکنش ها واکنش های تعادلی هستند که در آنها علاوه بر تشکیل محصولات واکنش از طریق واکنش رو به جلو، مواد اولیه نیز از محصولات واکنش از طریق واکنش معکوس تشکیل می شوند:
- |�آ|آ+|�ب|ب⇌|�ک|ک+|�L|L

اگر واکنش رو به جلو دارای آنتالپی آزاد مولی فعال باشدΔجیاچمنn‡و همچنین آنتالپی آزاد مولی واکنشΔآرجی

- |�آ|آ+|�ب|ب⟶|�ک|ک+|�L|L

و واکنش معکوس با آنتالپی آزاد مولی فعال سازیΔجیrتوهجک‡
- |�ک|ک+|�L|L⟶|�آ|آ+|�ب|ب

دقیقاً در امتداد همان مسیر واکنش در جهات مخالف برای آنتالپی آزاد مولی فعال سازی اعمال می شود.Δجیاچمنn‡واکنش رو به جلو:
- Δجیاچمنn‡=Δآرجی+Δجیrتوهجک‡

ثابت سرعت واکنش رو به جلوکاچمنnسپس خواهد شد:
 سپس خواهد شد:
سپس خواهد شد:- کاچمنn=ثابت⋅انقضا[-Δجیاچمنn‡آرتی]=ثابت⋅انقضا[-Δآرجی+Δجیrتوهجک‡آرتی]

برای ضریب k back و k back به صورت زیر است:
- کاچمنnکrتوهجک=انقضا[-Δآرجی+Δجیrتوهجک‡آرتی]⋅انقضا[-Δجیrتوهجک‡آرتی]-1

نتایج ساده سازی در:
- کاچمنnکrتوهجک=انقضا[-Δآرجی+Δجیrتوهجک‡آرتی]⋅انقضا[Δجیrتوهجک‡آرتی]=انقضا[-Δآرجی-Δجیrتوهجک‡+Δجیrتوهجک‡آرتی]

بدین ترتیب تبدیل می شود:
- کاچمنnکrتوهجک=انقضا[-Δآرجیآرتی]=ک

وجود داردکثابت تعادل ترمودینامیکی واکنش مورد بررسی. ثابت های نرخکاچمنnواکنش رو به جلو وکrتوهجکبنابراین واکنش معکوس کامل استΔآرجیهمراه با یکدیگر – رابطهکاچمنnکrتوهجکبا ثابت تعادل ترمودینامیکی تعیین می شود. این ارتباط اغلب به این معناست که ثابت تعادل یک واکنش تعادلی به ثابت سرعت واکنش های رو به جلو و معکوس بستگی دارد. با این حال، این ایده بر اساس یک پیش فرض نادرست است . کمیتهای ترمودینامیکی که تغییرات حالت را توصیف میکنند، مانند آنتالپی آزاد واکنش و ثابت تعادل، منحصراً به حالتهای اولیه و نهایی بستگی دارند، اما نه به مسیری که در طول آن سیستم از حالت اولیه به حالت نهایی حرکت میکند.
 ثابت تعادل ترمودینامیکی واکنش مورد بررسی. ثابت های نرخکاچمنn
ثابت تعادل ترمودینامیکی واکنش مورد بررسی. ثابت های نرخکاچمنn بنابراین واکنش معکوس کامل استΔآرجی
بنابراین واکنش معکوس کامل استΔآرجی همراه با یکدیگر – رابطهکاچمنnکrتوهجک
همراه با یکدیگر – رابطهکاچمنnکrتوهجک با ثابت تعادل ترمودینامیکی تعیین می شود. این ارتباط اغلب به این معناست که ثابت تعادل یک واکنش تعادلی به ثابت سرعت واکنش های رو به جلو و معکوس بستگی دارد. با این حال، این ایده بر اساس یک پیش فرض نادرست است . کمیتهای ترمودینامیکی که تغییرات حالت را توصیف میکنند، مانند آنتالپی آزاد واکنش و ثابت تعادل، منحصراً به حالتهای اولیه و نهایی بستگی دارند، اما نه به مسیری که در طول آن سیستم از حالت اولیه به حالت نهایی حرکت میکند.
با ثابت تعادل ترمودینامیکی تعیین می شود. این ارتباط اغلب به این معناست که ثابت تعادل یک واکنش تعادلی به ثابت سرعت واکنش های رو به جلو و معکوس بستگی دارد. با این حال، این ایده بر اساس یک پیش فرض نادرست است . کمیتهای ترمودینامیکی که تغییرات حالت را توصیف میکنند، مانند آنتالپی آزاد واکنش و ثابت تعادل، منحصراً به حالتهای اولیه و نهایی بستگی دارند، اما نه به مسیری که در طول آن سیستم از حالت اولیه به حالت نهایی حرکت میکند.انرژی های فعال سازی

وجود موانع بالقوه ای که باید در جریان یک واکنش ناخالص قابل مشاهده پدیدارشناسی در هنگام تبدیل مواد اولیه به محصولات غلبه کرد، به طور تجربی با معادله آرنیوس نشان داده می شود. بر خلاف معادله آیرینگ که از تئوری حالت گذار ناشی می شود، معادله آرنیوس تعداد یا ماهیت حالت های انتقالی را که باید توسط سیستم واکنش دهنده عبور کند را در نظر نمی گیرد، بلکه موانع بالقوه ای را نشان می دهد که باید بر آن غلبه کرد. از طریق پارامتر پدیدارشناسی انرژی فعال سازی�آ. اگر انرژی فعال سازی در دمای ثابت به بی نهایت نزدیک شود، ثابت سرعت مطابق معادله آرنیوس به صفر همگرا می شود. اگر انرژی فعال سازی صفر باشد، ثابت سرعت برابر با ضریب پیش نمایی می شودآ. به طور مشابه، معادله آرنیوس نیز می تواند برای نشان دادن نیمه عمر استفاده شودتی1/2به انرژی فعال سازی بستگی دارد. اگر انرژی فعال سازی در دمای ثابت به بی نهایت نزدیک شود، نیمه عمر نیز به بی نهایت نزدیک می شود. اگر انرژی فعال سازی صفر باشد، نیمه عمر برابر با ضریب پیش نمایی می شودآ”.
به طور کلی، در دمای ثابت، با کاهش انرژی فعالسازی، سرعت واکنش افزایش مییابد. بنابراین هنگام کاتالیز واکنش های شیمیایی، مسیرهای واکنش جایگزین ارائه می شود که منجر به کاهش انرژی فعال سازی می شود. این به سرعت واکنش بالاتری بدون نیاز به افزایش دمای واکنش دست می یابد.
پروفیل های غلظت-زمان برای یک واکنش در چندین دما به دست می آیند و ثابت های سرعت مربوطه از آنها به دست می آیند.ک(تی)انرژی فعال سازی را می توان به صورت تجربی نیز تعیین کرد�آتعیین کنید. لگاریتم کردن معادله آرنیوس را به یک معادله خط مستقیم تبدیل می کند :
 انرژی فعال سازی را می توان به صورت تجربی نیز تعیین کرد�آ
انرژی فعال سازی را می توان به صورت تجربی نیز تعیین کرد�آ- لوگاریتمک=لوگاریتمآ-�آآر⋅1تی

نمودار گرافیکی لگاریتم طبیعیلوگاریتمک(تی)ثابت های سرعت تعیین شده تجربیک(تی)در برابر1/(آر⋅تی)منجر به یک خط مستقیم می شود که شیب آن انرژی فعال سازی منفی است-�آمطابقت دارد. تقاطع خط باyمحور لگاریتم استلوگاریتمآاز عامل پیش نماییآمعادله آرنیوس
 ثابت های سرعت تعیین شده تجربیک(تی)
ثابت های سرعت تعیین شده تجربیک(تی) منجر به یک خط مستقیم می شود که شیب آن انرژی فعال سازی منفی است-�آ
منجر به یک خط مستقیم می شود که شیب آن انرژی فعال سازی منفی است-�آ مطابقت دارد. تقاطع خط باy
مطابقت دارد. تقاطع خط باy محور لگاریتم استلوگاریتمآ
محور لگاریتم استلوگاریتمآ از عامل پیش نماییآ
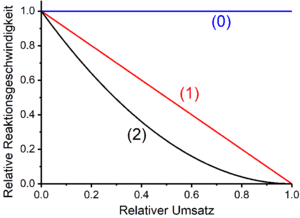
از عامل پیش نماییآواکنش های مرتبه صفر تا سوم
 .
.در بسیاری از موارد، سرعت واکنش های شیمیایی دخیل استنمواد اولیهز�متناسب با محصولات توابع توان غلظت مواد اولیه درگیر هستند[ز�]:
 مواد اولیهز�
مواد اولیهز� متناسب با محصولات توابع توان غلظت مواد اولیه درگیر هستند[ز�]
متناسب با محصولات توابع توان غلظت مواد اولیه درگیر هستند[ز�] :
:- �=ک∏�=1ن[ز�]به عنوان مثال�

اینجاستکنرخ ثابت شارحانبه عنوان مثال�دستورات واکنش جزئی در رابطه با مواد اولیه هستندز�. مجموع∑�=1نبه عنوان مثال�ترتیب کلی واکنش واکنش مورد بررسی است. دستورات واکنش جزئی می توانند، اما مجبور نیستند، مقادیر مشابهی با اعداد استوکیومتری مواد اولیه مربوطه در یک واکنش داشته باشند. اعداد استوکیومتری که در معادلات واکنش ناخالص ظاهر می شوند��مواد اولیهز�اغلب نشان دهنده استوکیومتری کلی واکنش های مرکب است که شامل چندین واکنش ابتدایی است. دستورات واکنش جزئی و در نتیجه ترتیبات واکنش کلی اغلب، مانند جانشینی ها و حذف های نوکلئوفیل مرتبه اول ، از واکنش های ابتدایی تعیین کننده سرعت که استوکیومتری آنها با استوکیومتری واکنش ناخالص مورد بررسی متفاوت است، منتج می شود. اگر یک واکنش ناخالص، مانند واکنشهای زنجیرهای رادیکال ، شامل واکنشهای ابتدایی همپوشانی یا جفتشده بدون وجود یک واکنش ابتدایی آهسته و در نتیجه تعیینکننده سرعت باشد، ترتیب واکنش جزئی نیز میتواند با اعداد استوکیومتری اجزای مورد نظر متفاوت باشد. در چنین مواردی، دستورات واکنش شکسته نیز رخ می دهد. [4] [5]
 نرخ ثابت شارحانبه عنوان مثال�
نرخ ثابت شارحانبه عنوان مثال� دستورات واکنش جزئی در رابطه با مواد اولیه هستندز�
دستورات واکنش جزئی در رابطه با مواد اولیه هستندز� ترتیب کلی واکنش واکنش مورد بررسی است. دستورات واکنش جزئی می توانند، اما مجبور نیستند، مقادیر مشابهی با اعداد استوکیومتری مواد اولیه مربوطه در یک واکنش داشته باشند. اعداد استوکیومتری که در معادلات واکنش ناخالص ظاهر می شوند��
ترتیب کلی واکنش واکنش مورد بررسی است. دستورات واکنش جزئی می توانند، اما مجبور نیستند، مقادیر مشابهی با اعداد استوکیومتری مواد اولیه مربوطه در یک واکنش داشته باشند. اعداد استوکیومتری که در معادلات واکنش ناخالص ظاهر می شوند�� مواد اولیهز�
مواد اولیهز�قوانین زمان واکنشها، که ترتیب کلی واکنش آنها را میتوان تعیین کرد، میتوانند به معادلات خط مستقیم تبدیل شوند ( جدول زیر را ببینید ؛ برای واکنشهای مرتبه صفر، پروفایلهای غلظت-زمان همیشه خطی هستند). قوانین زمان ارائه شده در این فرم را می توان برای تعیین تجربی ترتیب کلی واکنش با غلظت های اندازه گیری شده پس از زمان های مختلف واکنش استفاده کرد.[آ](تی)یک واکنش دهندهآمقایسه شود. مقدار شیب خط مستقیم به دست آمده از این طریق با حاصل ضرب عدد استوکیومتری مطابقت دارد.�آاز مواد اولیهآو نرخ ثابتک.
 از مواد اولیهآ
از مواد اولیهآنمای کلی
قوانین زمان، نمودارهای خطی قوانین زمان برای تعیین ثابت سرعتکو عبارات نیمه عمر برای واکنش هایی با قوانین نرخ از نوع هستند�=ک[آ]�در جدول زیر با در نظر گرفتن عدد استوکیومتری�آاز مواد اولیهآخلاصه شده است. [6] [7] لازم به ذکر است که�آعلامت منفی دارد واحدهای ثابت سرعتکبرای همه واکنش ها با ترتیب کلی واکنش هستند�معتبر.
 در جدول زیر با در نظر گرفتن عدد استوکیومتری�آ
در جدول زیر با در نظر گرفتن عدد استوکیومتری�آ خلاصه شده است. [6] [7] لازم به ذکر است که�آ
خلاصه شده است. [6] [7] لازم به ذکر است که�آ معتبر.
معتبر.| 0. سفارش دهید | سفارش 1 | سفارش 2 | n _ سفارش (�≠1) | |
|---|---|---|---|---|
| قانون سرعت | 1/�آ⋅د[آ]/دتی=ک | 1/�آ⋅د[آ]/دتی=ک[آ] | 1/�آ⋅د[آ]/دتی=ک[آ]2 | 1/�آ⋅د[آ]/دتی=ک[آ]� |
| قانون زمان | [آ]=[آ]0+�آکتی | [آ]=[آ]0ه�آکتی | [آ]=1/(1[آ]0-�آکتی) | 1[آ]�-1=1[آ]0�-1-(�-1)�آکتی |
| واحدک | مترOلس⋅متر3 | 1س | متر3مترOل⋅س | متر3�-3مترOل�-1⋅س |
| طرح خطی قانون زمان | [آ]در مقابل تی | لوگاریتم([آ][آ]0)در مقابل تی | 1[آ]در مقابل تی | 1[آ]�-1در مقابل تی |
| نیمه عمر | تی1/2=-[آ]02�آک | تی1/2=-لوگاریتم(2)�آک | تی1/2=-1�آک[آ]0 | تی1/2=-2�-1-1(�-1)�آک[آ]0�-1 |
 )
)











 در مقابل تی
در مقابل تی در مقابل تی
در مقابل تی در مقابل تی
در مقابل تی در مقابل تی
در مقابل تی



واکنش های مرتبه صفر
سرعت واکنش های مرتبه صفر مستقل از غلظت واکنش دهنده ها است. یعنی سرعت واکنش ثابت است. بنابراین قوانین زمان به شکل یک خط مستقیم با شیب منفی (برای متغیرهای فروش و واکنش دهنده ها) یا با شیب مثبت (برای محصولات) هستند. شیب خط مطابق با بزرگی ثابت سرعت k است . نمونه هایی از واکنش های مرتبه صفر شامل واکنش های فتوشیمیایی و کاتالیزوری خاص است . به عنوان مثال، اکسیداسیون بیولوژیکی اتانول به استالدئید توسط الکل دهیدروژنازهای خاص نسبت به اتانول درجه صفر است. [8] مثال دیگر پلیمریزاسیون امولسیونی است که در آن پس از مرحله شروع واکنش درجه صفر است. [9]
واکنش های مرتبه اول
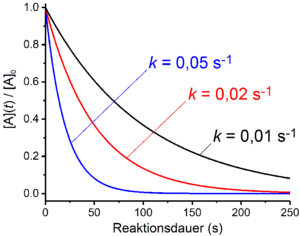
 از یک ماده اولیهآ
از یک ماده اولیهآدر واکنش های مرتبه اول، سرعت واکنش به صورت خطی به غلظت یک واکنش دهنده A بستگی دارد. این در صورتی است که فرآیند کلی فقط شامل یک واکنش فروپاشی تک مولکولی باشد . نمونه ای از این واپاشی رادیواکتیو است . علاوه بر این، واکنشهایی که شامل چندین واکنش ابتدایی میشوند، اگر مرحله تعیینکننده سرعت یک فرآیند فروپاشی یا تفکیک باشد، مرتبه اول هستند . نمونه هایی از این مکانیسم SN 1 جایگزینی نوکلئوفیل یا مکانیسم E1 در واکنش های حذف است . قانون نرخ برای یک واکنش مرتبه اول به شکل زیر است:
- �=1�آ⋅د[آ]دتی=ک⋅[آ]

قانون زمان مرتبه اول با جدا کردن متغیرهای زیر به دست می آید:
- 1�آ⋅1[آ]⋅د[آ]=ک⋅دتی

مرحله بعدی محاسبه انتگرال های معین است:
- 1�آ⋅🔻[آ]0[آ](تی)1[آ]⋅د[آ]=🔻0تیک⋅دتی

ابتدا با در نظر گرفتن این واقعیت کهلوگاریتم[آ]تابع ضد مشتق1/[آ]است:
 تابع ضد مشتق1/[آ]
تابع ضد مشتق1/[آ] است:
است:1�آ⋅{لوگاریتم[آ](تی)-لوگاریتم[آ]0}=ک⋅تی

اعمال قواعد لگاریتمی منجر به موارد زیر می شود:
- 1�آ⋅لوگاریتم[آ](تی)[آ]0=ک⋅تی

نتایج تغییر شکل در:
- لوگاریتم[آ](تی)[آ]0=�آ⋅ک⋅تی

دیلوگاریتم کردن قانون زمان را برای واکنش های مرتبه اول می دهد:
- [آ](تی)[آ]0=ه�آ⋅ک⋅تی

به ترتیب:
- [آ](تی)=[آ]0⋅ه�آ⋅ک⋅تی

اگر، همانطور که اغلب در مورد واکنش های مرتبه اول اتفاق می افتد،
- �آ=-1،
 ،
،قانون زمان برای واکنش های مرتبه اول تبدیل می شود:
- [آ](تی)=[آ]0⋅ه-ک⋅تی

واکنش های مرتبه دوم
مرتبه دوم واکنش های ابتدایی بر اساس برخوردهای دو مولکولی هستند. نمونه هایی از این جانشینی های هسته دوست با استفاده از مکانیسم S N 2 و حذف با استفاده از مکانیسم E2 هستند. اگر فرآیندهای فروپاشی یا تفکیک مرتبه اول نیز در جریان یک واکنش چند مرحلهای رخ دهد، این مراحل معمولاً مراحل تعیینکننده سرعت هستند، به طوری که مانند واکنشهای SN1 و E1، واکنش کلی مرتبه اول است.
برخورد دو مولکولی مربوط به دو ذره مشابه از گونه را پیدا می کندآدر عوض، مقدار عدد استوکیومتری است�آاز مواد اولیهآمعادل دو است و معادله واکنش به شکل زیر است:
 از مواد اولیهآ
از مواد اولیهآ- 2آ⟶محصولات

قوانین سرعت و زمان حاصل و همچنین بیان نیمه عمر[آ](تی)در جدول بالا ذکر شده اند .
در برخورد دو مولکولی ذرات دو گونه مختلف وجود داردآوبدرگیر هستند و مقادیر اعداد استوکیومتری هستند�آو�بواکنش دهنده هاآوبهر کدام برابر با یک، معادله واکنش به صورت زیر می شود:
 واکنش دهنده هاآ
واکنش دهنده هاآ- آ+ب⟶محصولات

قانون سرعت بر این اساس تبدیل می شود:
- �=-د[آ]دتی=-د[ب]دتی=ک⋅[آ]⋅[ب]

به عنوان یک قانون زمان شما با[ب]0به عنوان غلظت اولیه واکنش دهندهبقبل از شروع واکنش: [10]
 به عنوان غلظت اولیه واکنش دهندهب
به عنوان غلظت اولیه واکنش دهندهب- [آ]=[آ]0⋅{[آ]0-[ب]0}⋅ه{[آ]0-[ب]0}کتی[آ]0⋅ه{[آ]0-[ب]0}کتی-[ب]0

واکنش های مرتبه سوم
برای اینکه یک واکنش درجه سوم باشد، باید یک برخورد سه مولکولی را به عنوان یک واکنش ابتدایی شامل شود. از آنجایی که احتمال برخورد سه مولکولی کم است، واکنش های مرتبه سوم نادر هستند. وقوع واکنش های مرتبه سوم گزارش شده است، به عنوان مثال، در ارتباط با واکنش های نوترکیبی اتم ها و رادیکال های نوع
- 2آ+م⟶آ2+م،
 ،
،که در آن M به عنوان یک شریک برخورد عمل می کند، و همچنین برای واکنش های مونوکسید نیتروژن با هالوژن X و اکسیژن مطابق با
- 2نه+O2⟶2نه2

و
- 2نه+ایکس2⟶2NOX

فرضیه ها [11]
قوانین سرعت واکنش های تعادلی
یک واکنش تعادلی در نظر گرفته می شود
- آ⇌ب.
 .
.اگر واکنش های رو به جلو و معکوس مرتبه اول باشند، تغییرات در غلظت هاآوبتوسط معادلات دیفرانسیل تعیین می شود
 توسط معادلات دیفرانسیل تعیین می شود
توسط معادلات دیفرانسیل تعیین می شود- �آ=د[آ]دتی=-کآب⋅[آ]+کبآ⋅[ب]
- �ب=د[ب]دتی=+کآب⋅[آ]-کبآ⋅[ب]


با ثابت های نرخکآبواکنشآ→بوکبآواکنش پشتب→آو همچنین شرایط
 واکنشآ→ب
واکنشآ→ب وکبآ
وکبآ واکنش پشتب→آ
واکنش پشتب→آ و همچنین شرایط
و همچنین شرایط- د[آ]+د[ب]=0.

برای حل این سیستم معادلات دیفرانسیل، شرایط مرزی زیر ضروری است:
- تی :[آ]=[آ]0-ایکس،[ب]=[ب]0+ایکس
- تی=∞ :[آ]هq=[آ]0-ایکسهq،[ب]هq=[ب]0+ایکسهq
 ،[ب]=[ب]0+ایکس
،[ب]=[ب]0+ایکس
 :[آ]هq=[آ]0-ایکسهq
:[آ]هq=[آ]0-ایکسهq ،[ب]هq=[ب]0+ایکسهq
،[ب]هq=[ب]0+ایکسهq
دو معادله دیفرانسیل برایآوببا این شرط مرزی ساده می شوند:
 با این شرط مرزی ساده می شوند:
با این شرط مرزی ساده می شوند:- دایکسدتی=(کآب+کبآ)(ایکسهq-ایکس)

این منجر به قانون زمان انتگرال زیر برای واکنش های تعادلی مرتبه اول می شود:
- لوگاریتمایکسهqایکسهq-ایکس=(کآب+کبآ)⋅تی

با
- [آ]هq=([آ]0+[ب]0)⋅کبآکآب+کبآو[ب]هq=([آ]0+[ب]0)⋅کآبکآب+کبآ
 و[ب]هq=([آ]0+[ب]0)⋅کآبکآب+کبآ
و[ب]هq=([آ]0+[ب]0)⋅کآبکآب+کبآ
همچنین
- [آ]0-[آ]هq=[ب]هq-[ب]0=کآب[آ]0-کبآ[ب]0کآب+کبآ

قوانین سرعت زیر نتیجه می شود:
- [آ]تی=([آ]0+[ب]0)⋅کبآکآب+کبآ+کآب[آ]0-کبآ[ب]0کآب+کبآ⋅ه-(کآب+کبآ)⋅تی

- [ب]تی=([آ]0+[ب]0)⋅کآبکآب+کبآ-کآب[آ]0-کبآ[ب]0کآب+کبآ⋅ه-(کآب+کبآ)⋅تی

اندازه گیری پروفیل غلظت-زمان
سینتیک واکنشهای شیمیایی با تعیین پروفیلهای غلظت-زمان با استفاده از روشهای تحلیلی کمی مورد بررسی قرار میگیرد. برای این منظور، شیمی تجزیه طیف گسترده ای از روش ها را ارائه می دهد که به دلیل پیشرفت فناوری تجزیه و تحلیل ابزاری و ریزواکنش، به طور مداوم در حال تکامل است . متغیر اندازه گیری انتخاب شده برای تعیین پروفایل غلظت-زمان باید از نظر کمی با غلظت اجزای مشاهده شده متناسب باشد. روشهای متداول برای تعیین پروفیلهای غلظت-زمان شامل اندازهگیری ثابت دی الکتریک ، ضریب شکست ، فعالیت نوری ، فلورسانس یا هدایت محلول واکنش، اندازهگیری تغییرات حجم یا فشار، کالریسنجی و همچنین جذب و انتشار است. طیف سنجی و پراکندگی نور .
متغیر اندازه گیری شده را می توان روی خود مخلوط واکنش بدون تماس و بدون نمونه برداری از مخلوط واکنش مشاهده کرد. این روش سودمند است زیرا اختلالات در فرآیند واکنش را به حداقل می رساند. روش دیگر مبتنی بر این است که مقدار کمی از مخلوط واکنش به طور منظم و معمولاً به طور خودکار گرفته می شود. اینها را می توان تحت روشهای تجزیه و تحلیل غیر مخرب قرار داد و سپس دوباره با مخلوط واکنش ترکیب کرد. همچنین می توان از روش های تجزیه و تحلیلی که با مصرف مقدار کمی از آن ها مرتبط است استفاده کرد. اگر مقدار کمی از مخلوط واکنش گرفته شود، مداخله بیشتری در واکنش نسبت به فرآیندهای غیر تماسی وجود دارد. با این حال، سودمند است که می توان از طیف وسیع تری از روش های تجزیه و تحلیل استفاده کرد و از روش های تجزیه و تحلیلی استفاده کرد که یا حساس تر هستند یا اطلاعات بیشتری نسبت به روش های تجزیه و تحلیلی که می توانند برای مشاهده غیر تماسی مخلوط واکنش استفاده شوند، ارائه می دهند. نمونه های استخراج شده نیز می توانند تحت تجزیه و تحلیل کیفی پیچیده تری قرار گیرند، برای مثال با جداسازی اجزای آنها با استفاده از کروماتوگرافی گازی یا کروماتوگرافی مایع با کارایی بالا قبل از بررسی تحلیلی واقعی. تحت شرایط خاص، واکنش در کسری های حذف شده باید کند یا متوقف شود تا با ادامه واکنش پس از نمونه برداری، از تحریف نتایج آنالیز جلوگیری شود. این را می توان با سرد کردن قوی مخلوط واکنش حذف شده یا با حذف یک جزء واکنشی از مخلوط واکنش، به عنوان مثال با رسوب انجام داد .
برای اندازهگیری پروفیلهای غلظت-زمان، واکنشدهندهها باید آنقدر سریع مخلوط شوند که زمان شروع مشخصی برای بررسی واکنش قابل شناسایی باشد. این را می توان با کوچک کردن تنظیمات آزمایشی مورد استفاده با استفاده از فناوری ریزواکنش به دست آورد، زیرا مسیرهای انتقال واکنش دهنده ها سپس کوتاه می شوند. برای واکنش های آهسته، مقادیر مشخصی از مواد را می توان با استفاده از همزن های ساده، لوله های جریان یا اتاق های اختلاط با دقت بالا مخلوط کرد. برای واکنشهای سریعتر، که با مقیاسهای زمانی در محدوده دقیقه تا ثانیه مشخص میشوند، اغلب از دستگاه جریان ویژه استفاده میشود. برای واکنشهای بسیار سریع، که با مقیاسهای زمانی در محدوده میلیثانیه مشخص میشوند، فرآیندهای بهینهسازی شده، مانند روش جریان متوقف شده ، برای اختلاط سریع و کارآمد مواد اولیه استفاده میشود.
گروه دوم از روشها برای مطالعه واکنشهای بسیار سریع با مقیاسهای زمانی مشخص تا محدوده پیکوثانیه، روشهای آرامسازی هستند . اینها بر اساس اصل مخلوط کردن مواد اولیه قبل از دوره مشاهده واقعی است. مخلوط واکنش شروع به واکنش می کند. پس از برقراری حالت تعادل در مخلوط واکنش، این حالت توسط یک شوک به سرعت اعمال شده مختل می شود و با استفاده از روش های تحلیلی مناسب، شل شدن مخلوط واکنش به حالت تعادل جدید بررسی می شود. نمونه هایی از فرآیندهای آرامش عبارتند از فتولیز فلاش و همچنین فرآیندهای دما، فشار و پرش میدانی.
تاریخچه سینتیک
اولین مطالعات کیفی در مورد سینتیک در اوایل سال 1777 توسط کارل فردریش ونزل در اثرش الهیات خویشاوندی اجسام منتشر شده در درسدن گزارش شد . بعدها، کلود لوئیس برتوله و ویلیام هیگینز نیز به سؤالات جنبشی پرداختند. [12] اولین کار واقعاً اساسی در مورد سینتیک، تقسیم قند نیشکر تحت تأثیر اسید، توسط لودویگ فردیناند ویلهلمی در سال 1850 ارائه شد. [13] Jacobus Henricus van’t Hoff صابونی شدن اتیل استات و هیدرولیز اسید کلرواستیک را در سال 1896 بررسی کرد. او معادلات سرعت واکنش ها را به صورت ریاضی فرموله کرد. او همچنین قوانین اساسی حاکم بر وابستگی دمایی سرعت واکنش را توسعه داد. [14] Svante Arrhenius اشتقاق را بهبود بخشید و هنگامی که دما به میزان 1 K به عنوان یک قانون کلی برای تغییر سرعت واکنش افزایش یافت، سرعت واکنش را حدود 12٪ افزایش داد (به قانون RGT مراجعه کنید ). FEC Scheffer و WF Brandsma انرژی استاندارد فعال سازی گیبس را برای ثابت سرعت در سال 1926 معرفی کردند. [15] مانفرد ایگن ، رونالد نوریش و جورج پورتر جایزه نوبل شیمی را در سال 1967 برای توسعه روش های آرام سازی برای مطالعه سینتیک واکنش های سریع دریافت کردند .